The following points highlight the six common types of DNA markers. The types are: 1. Single-nucleotide Polymorphism 2. Short Tandem Repeats 3. Variable Number Tandem Repeats 4. Restriction Fragment Length Polymorphism 5. Randomly Amplified Polymorpohic DNA 6. Amplified Fragment Length Polymorphism.
Type # 1. Single-nucleotide Polymorphism (SNPs or “Snips”):
The single-nucleotide polymorphism is a single base-pair change or a point mutation at a SNP locus or site (Fig. 9.46). It is the most common type of DNA polymorphism occurring at a frequency of about 1 per 350 bp. It accounts about 90-95 percent of DNA sequence variation.

Most of the SNPs occur in non-coding regions of the genome and are called non-coding SNPs while SNPs occurring in coding regions are known as cSNPs or coding SNPs. In human, each gene has about four cSNPs, half of which cause missense mutations in the encoded protein and half of which cause silent mutations. Many missense mutations caused by cSNPs result in genetic diseases in human.
ADVERTISEMENTS:
Typing of SNPs:
SNPs can be detected by following methods:
(i) Oligonucleotide hybridisation analysis:
An oligonucleotide is a short, single- stranded DNA molecule, usually less than 50 nucleotides in length, synthesised in test tube. In this process, such oligonucleotide is mixed with target DNA and allowed to hybridise.
ADVERTISEMENTS:
If target DNA has common allele with the oligonucleotide, then hybridisation occurs. But if there is a single mismatch – a single position within the oligonucleotide that does not form a base pair – then hybridisation does not occur (Fig. 9.47A). Oligonucleotide hybridisation can therefore discriminate between two alleles of an SNP.

(ii) DNA chip:
This technology uses a wafer of glass or silicon, 2.0 cm2 or less in area; carrying many different oligonucleotides in a high density array. The DNA to be tested is labeled with a fluorescent marker and then pipetted onto the surface of the chip.
ADVERTISEMENTS:
Hybridisation occurs where oligonucleotides find complementary DNA strands. This hybridisation is then detected by examining the chip with a fluorescence microscope, the position from which the fluorescent signal is emitted indicating that oligonucleotides have hybridized with the test NDA. Many SNPs can, therefore, be scored in a single experiment.
(iii) Solution hybridisation techniques:
It is carried out in a wells of a microtiter tray, each well containing different oligonucleotides. This technique uses several detection system that can discriminate between hybridised, single-stranded DNA and double-stranded product that results when an oligonucleotide hybridizes to the test DNA.
One such system uses a pair of labels comprising a fluorescent dye and a compound that quenches the fluorescent signal when brought into close proximity with the dye. The dye is attached to one end of an oligonucleotide and the quenching compound to the other end.
Normally there is no fluorescence because the oligonucleotide is designed in such a way that the two ends base pair to one another, placing the quencher next to the dye (Fig. 9.47B). Hybridisation between oligonucleotide and test DNA disrupts this base pairing, moving the quencher away from the dye and enabling the fluorescent signal to be generated and detected.
Type # 2. Short Tandem Repeats (STRs):
These are 2-6 bp long DNA sequences repeated tandemly. STRs are also called microsatellites and simple sequence repeats (SSRs). One STR may be repeated up to 100 times. In human genome, the number of dinucleotide repeats is 128,000, trinucleotide repeat is 8,740, four nucleotide repeat is 23,680, five nucleotide repeat is 4,300, and sex nucleotide repeat is 230.
In a population, most of the STRs are polymorphic, so they can be used to study genetic mapping and in forensics. PCR is used to determine such polymorphic STRs present in the genome. For example, PCR with primers that flank the STR locus will produce different length DNA fragments consisting of the STR span plus the DNA from the STR to the leftmost end of the left primer and to the rightmost end of the right primer.
One particular human STR locus has a dinucleotide repeat GTTA. This allele varies from 6 to 15 copies in number. PCR of such locus from two alleles will generate DNA fragments of different length (Fig. 9.48) which can be visualised by agarose gel electrophoresis. Thus, with the help of PCR, STRs can be used as a marker in distinguishing the genomic DNA from different individuals.

Type # 3. Variable Number Tandem Repeats (VNTRs):
VNTR was the first DNA sequence polymorphism that was demonstrated in human genome. In 1985, Alec J. Jeffreys described such DNA polymorphism which is also called minisatellites. These are similar to STRs but VNTR repeat lengths are longer than those in STRs, from 75 to few tens of base pairs long. For example, the base sequence GGAAGGGAAGGGAAGGGAAG is composed of four tandem repeats of the five nucleotide sequence GGAAG.
ADVERTISEMENTS:
Clusters of such sequences are widely dispersed in the human genome. The number of repeats at each locus ranges from 2 to more than 100. The number of repeats at a given locus is variable, and each variation constitutes a VNTR allele. Most loci have dozens of alleles each, as a result, heterozygosity is common. In human genome, the number of VNTR loci is less than STR loci.
Because of their longer length than STR, not the PCR, but restriction digestion and Southern blot are used to study the VNTRs in a genome. For restriction digestion, the genomic DNA is isolated and then cut with a restriction enzyme that cuts on either side of the VNTR locus.
The restriction fragments are then separated by gel electrophoresis and transferred to a membrane filter by Southern blotting. With the help of a probe for a particular repeat sequence of the VNTR locus, the total length of that VNTR locus could be determined.
Two types of VNTR loci have been found in an organism’s genome – unique loci and multi-copy loci. If a probe detects only one VNTR locus, it is called a monolocus or single-locus probe. Probes that detect VN FR loci at a number of sites in the genome are called multilocus probes. In an organism’s genome unique loci is present in a single copy but in case of multi-copy loci there may be a number of copies scattered around the genome.
Type # 4. Restriction Fragment Length Polymorphism (RFLP):
In a population of individuals belonging to one or more species or cells, often there is a variable region in the genomic DNA. If one nucleotide sequence variation occurs in this region, it results in alteration in the pattern of DNA fragments generated after digestion with a restriction endonuclease, thus an RFLP is generated. For example, the restriction endonuclease EcoRI cut double-stranded DNA at
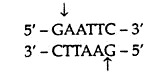
and not at other sequence. This sequence specificity means that treatment of a DNA molecule with this enzyme should always produce same set of fragments. But this is not always the case with genomic DNA because some restriction sites are polymorphic, existing as two following alleles:
(i) Allele displaying the correct sequence for the restriction site and, therefore, being cut when the DNA is treated with the enzyme,
(ii) Allele with a sequence alteration so the restriction site is no longer recognised. This is the basis of existence of RFLP. Variation in one DNA fragment obtained with specific restriction enzyme is treated as one RFLP. RFLP is usually detected by restriction digestion coupled with Southern hybridisation where a specific DNA probe is used to identify the genomic region. Hence, RFLPs are the representative of the genotype and therefore, they are used as molecular markers.
Detection of RFLPs:
In order to demonstrate the RFLPs, it is necessary to determine the size of individual restriction fragments.
It can be achieved through the following steps:
(a) First the DNAs from several individuals are isolated from the same tissues and then purified.
(b) The DNAs are then digested with the same restriction endonuclease separately.
(c) Each DNA digest is subjected to gel electrophoresis separately, but on the same gel slab.
(d) The DNA fragments obtained in the gel following electrophoresis usually form continuous smear because the number of fragments is large and the range in size is rather continuous. To visualize the fragments clearly, electrophoresis is, therefore, followed by Southern blotting.
Following electrophoresis, the test DNA fragments are denatured in strong alkali. The DNA fragments are then transferred by blotting on to a durable nitrocellulose or nylon membrane, to which single stranded DNA binds readily. The individual DNA fragments become immobilised on the membrane at positions which are a faithful record of the size separation achieved by gel electrophoresis.
Subsequently, the immobilised single-stranded target DNA sequences are allowed to associate with labeled single- stranded probe DNA. This probe will bind only to related DNA sequences in the target DNA and their position on the membrane can be related back to the original gel in order to estimate their size. The DNA fragments hybridizing with the probe, can be visualised by autoradiography, and will be called RFLPs at the phenotypic level.
Uses of RFLP:
(i) For evolutionary studies:
In a set of related species of plant or animal, restriction fragment length polymorphisms can be studied using a random or a specific DNA probe. The similarities or dissimilarities can then be used to establish the phylogenetic relationship among them.
(ii) For chromosome mapping:
In a number of species, RFLPs have been used to prepare chromosome maps. If a large number of RFLPs are available in a species, the parents, F1 and F2s can be used to study their inheritance and linkage relationship and genetic linkage maps can be prepared. A difference in restriction maps between two individuals or a RFLP can be used as a genetic marker like other phenotypic markers.
For example, presence of an additional site will produce two fragments and its absence will produce only one fragment making a difference that can be revealed by gel electrophoresis (Fig. 9.49). If there is a linkage between a molecular marker and a phenotypic trait, the change in molecular maps (restriction map) may not affect the phenotype. However, a tight linkage between molecular marker and the phenotypic marker sometimes may allow identification of genetic loci at molecular level.

(iii) Detection of pathogenic point mutation:
Occasionally, pathogenic mutations may abolish or create a restriction site, enabling direct screening of the pathogenic mutation. For example, in sickle cell mutation, a single nucleotide substitution (A → T) occurs at codon G in the β-gobin gene, which causes a missense mutation (Glu → Val).
It also eliminates a cutting site for the enzymes Mstll and Cunl. As a result, this mutation alters the pattern of restriction fragments seen on Southern blots. This characteristic RFLP patterns can be used for the prenatal diagnosis of sickle-cell anaemia. For this purpose, DNA is extracted from fetal cells and digested with enzymes like Mstll.
This enzyme cuts thrice in the region of normal β-globin gene and produces two small DNA fragments (Fig. 9.50, column 3). In the mutant alele, the second Mstll site is lost, thus one large fragment is produced (Fig. 9.50, column 4). The restriction-digested DNA fragments can be separated by gel electrophoresis and then visualised by Southern blot hybridisation.

Thus RFLPs can also be used to detect many other genetic diseases.
Type # 5. Randomly Amplified Polymorpohic DNA (RAPD):
PCR provides selective amplification of a specific DNA segment. It needs information about target DNA sequences, so that oligonucleotides can be synthesised for their use as primers at the two ends of the DNA segment to be amplified. If the target DNA sequence is not known, then primers with arbitrary sequences can be used for amplification.
Such primers are called random primers. This PCR products provide a truly random sample of DNA markers which are described as randomly amplified polymorphic DNAs (RAPDs—pronounced as ‘rapids’). RAPDs have several advantages over RFLPs and can be used for different studies.
Detection of RAPDs:
In the conventional PCR, two sequence specific primers, derived from the complementary DNA strands with antiparallel orientation, are used to amplify the given DNA sequences flanked by the two primers. In random amplification of polymorphic DNA, genomic DNA is amplified by a PCR that uses single primer of arbitrary nucleotide sequence.
The nucleotide sequence of each primer is chosen within the constraints that the primer is nine or ten nucleotides in length, between 50-80% G + C in composition and contains no palindromes. From this RAPD-PCR, randomly amplified polymorphic DNAs can be identified and selected, though not all RAPD-PCR products are polymorphic. These RAPDs can easily be identified by agarose gel electrophoresis.
Applications of RAPDs:
(i) Randomly amplified polymorphic DNA is extensively used by scientists to construct purpose-oriented maps from plant genome, because RAPD can provide a large number of molecular markers and they can be easily identified by agarose gel electrophoresis.
(ii) RAPD has been successfully used to construct a genetic linkage map of the haploid zebra fish, a popular model system to study developmental biology.
(iii) RAPD may be combined with RFLP to increase the efficiency of molecular markers for genetic analysis. Being polymorphic, a RAPD marker may be labeled and used as a probe for RFLPs analysis, thus can eliminate the need for recombinant DNA cloning of probes in bacteria.
Advantages of RAPD over RFLP:
(i) In RAPD, some primers with arbitrary sequences are used for different species. So no species specific probes are required for different species as required in RFLPs. For example, primers used in RAPD-PCR in maize, can also be used for soya bean, human, Neurospora etc.
(ii) Generation of RAPD data is about 5 times quicker than RFLPs, since RAPD-PCR involves less number of steps to visualize the final products.
(iii) RAPD can use DNA of crude quality in nanogram quantity, whereas RFLP requires relatively pure quality DNA in microgram quantity.
Disadvantages of RAPD over RFLP:
However, RAPDs have two disadvantages also. First, the amplification of a specific sequence is sensitive to PCR condition, including template concentration, and so it can be difficult to correlate results obtained by different workers. A second limitation of RAPD method is that usually it cannot distinguish heterozygotes from one of the two homozygous genotypes.
To overcome these problems, a different method has been described by Konieczny and Ausubet (1993). In this method, sequence- tagged sites (STSs) are derived from genes that have already been mapped and sequenced. Where possible the primers used are chosen such that the PCR products include introns to maximise the possibility of finding polymorphisms.
The primary PCR products are subjected to digestion with a panel of restriction endonucleases until a polymorphism is detected. Such markers are called cleaved amplified polymorphic sequences (CAPS). It should be noted that RFLPs are well-suited to mapping newly cloned DNA sequences. They are not convenient to use for mapping genes, such as plant genes, which are the first identity of mutation. CAPS are much more useful for this purpose.
Type # 6. Amplified Fragment Length Polymorphism (AFLP):
Amplified fragment length polymorphism is a diagnostic fingerprinting technique that detects genomic restriction fragments and, therefore, it resembles the RFLP technique. However, it detects presence or absence of restriction fragments and not length differences.
The AFLP technique is particularly powerful because it requires no previous sequence characterisation of the target genome. It is widely used in mapping plant, bacterial and even viral genomes, but not in animal genomes, because it is dependent on the presence of high rates of substitutional variation in the DNA.
Detection of AFLP:
To prepare an AFLP template, genomic DNA is isolated and digested simultaneously with two restriction endonucleases, EcoRI and Msel. The former has a 6-base pair recognition site and the latter a 4 base pair recognition site. When used together, these enzymes generate small DNA fragments that will amplify well and are in the optimal size range (< 1kb) for separation on denaturing poly- acrylamide gels.
Following heat inactivation of the restriction enzymes, the genomic DNA fragments are ligated to EcoRI and Msel adaptors to generate template DNA for amplification. These common adaptor sequences flanking variable genomic DNA sequences serve as primer binding sites on the restriction fragments. Following this strategy, it is possible to amplify many DNA fragments without having prior sequence knowledge.
In AFLP technique, PCR is performed in two consecutive reactions. Firstly, during the pre-amplification reaction, genomic fragments are amplified with AFLP primers each having one selective nucleotide (Fig. 9.51). Secondly, the PCR products of this reaction are then diluted and used as a template for the selective amplification using two new AFLP primers which have two or three selective nucleotides.
In addition EcoRI selective primer is radiolabeled. After the selective amplification the PCR products are separated on a gel and the resulting DNA fingerprint is detected by autoradiography.

Applications of AFLP:
(i) The AFLP technique can generate fingerprints of any DNA regardless of the origin or complexity. However, the number of amplified fragments can be controlled by the cleavage frequency of the rare cutter enzyme and the number of selective bases.
In addition, the nature of amplified bonds may be controlled by the nature of the selective bases. Selective extension with rare di- or trinucleotides will result in reduction of the number of amplified fragments.
(ii) AFLP can bridge the gap between genetic and physical maps. Most AFLP fragments correspond to unique positions on the genome and, hence, can be exploited as landmarks.
(iii) AFLP can be used to generate high- density maps in plants.
(iv) It can also be used for fingerprinting cloned DNA segments. By using no or few selective nucleotides, restriction fragment fingerprints will be produced which subsequently can be used to line up individual clones and make contigs.