The following points highlight the top six types of mutation in gene. The types are: 1. According to Size and Quality 2. According to Direction 3. According to Magnitude and Phenotypic Effect 4. According to Origin 5. According to Cell Type 6. Other Types of Mutation.
Types of Mutation in Gene:
- According to Size and Quality
- According to Direction
- According to Magnitude and Phenotypic Effect
- According to Origin
- According to Cell Type
- Other Types of Mutation
Type # 1. According to Size and Quality:
A. Gene mutation and B. Chromosomal Mutation.
ADVERTISEMENTS:
A. Gene Mutation:
Mutation concerned with changes occurring within a single gene is called point or gene mutation. Gene mutations serve as the source of most new alleles and thus are the origin of much genetic variability within a population.
Gene mutation is generally caused by two ways:
(a) Base Substitution:
ADVERTISEMENTS:
When one base pair is replaced by another base pair, the changes are referred to as substitution.
Two formal terms are often used to describe nucleotide substitutions:
(i) Transition mutation involving a change from one purine to another purine or one pyrimidine to another pyrimidine. The four types of transitions are A ↔ G or C ↔ T;
(ii) Trans-version mutation when the base pair substitution involves a change from a purine to pyrimidine or a pyrimidine to purine. The eight types of transversitions are A ↔ T, G ↔ C, A ↔ C and G ↔ T (Fig. 3.75a, b).

Causes of Base Pair Substitution:
1. Tautomeric Shift:
In a stable DNA, the nitrogenous bases of the molecule remain in keto (C=0) or in amino (-NH2) forms. Guanine and thymine are found in keto form while adenine and cytosine remain in amino form. Watson and Crick recognized that the bases can also exist in alternate chemical forms, called tautomer’s (or structural isomers), each differing by only a single proton shift in the molecule.
The biologically important tautomer’s are the keto-enol form of thymine and guanine, and amino-imino forms of cytosine and adenine. Because of such shifting or tautomeric shift, changes-in the bonding structure of the molecule could result in base pair changes or point mutations.
For example, normal adenine has an NH2 group that provides a hydrogen atom for bonding with the complementary keto group of thymine. However, due to a tautomeric shift, the imino form of adenine will now bond in complementary fashion with cytosine (Fig. 3.76a, b).
Similarly, a tautomeric shift may occur in thymine, changing it from the keto form to the rare enol (COH) form that can now bond with guanine (Fig. 3.76b). Tautomeric shift can also result in trans version, in which a nucleotide base position occupied by a purine such as adenine, may be changed into a pyrimidine such as cytosine.
Such errors produced by tautomeric shift will appear in DNA molecules following replication and they are classified as copy errors. All these simple changes as a result of tautomeric shift may have far-reaching mutational consequences.

2. Base Analogue:
ADVERTISEMENTS:
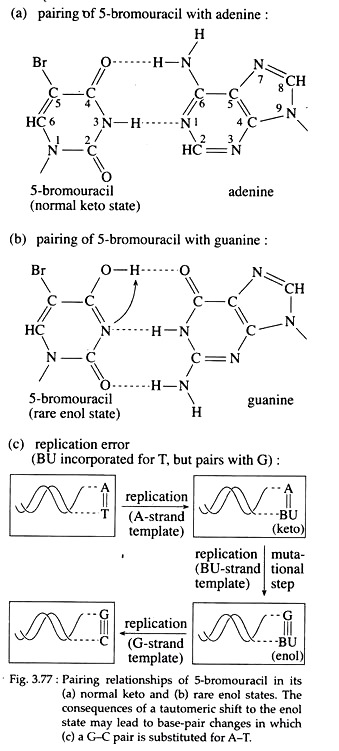
Molecules that may substitute for purines or pyrimidine’s during nucleic acid biosynthesis, are called base analogues and are often mutagenic chemicals. For example, 5-bromouracil (5-BU) a derivative of uracil, behaves as a thymine analogue and is halogenated at the 5′ position of the pyrimidine ring.
Now, if 5-BU is incorporated into DNA in place of thymine and a tautomeric shift to the endl forms occur, 5-BU then base pairs with guanine (Fig. 3.77a, b). So after one round of replication, an A=T to GsC transition results (Fig; 3.77c).
Furthermore, the presence of 5-BU within DNA increases its sensitivity to ultraviolet (UV) light, which itself is mutagenic. Examples of other base analogue is 2-amino purine (2-AP), an analogue of adenine.
3. Deamination:
Direct changes in nucleotide structure may be produced by agents like nitrous acid (HN02) that causes removal of amino group (deamination) from adenine and cytosine, thereby converting these bases to hypoxanthine (H) and uracil, respectively (Fig. 3.78 a, b).

This H can now pair with C and U with A. So, further replications lead to transition in which G is substituted for A and T for C. Deamination of guanine also occurs with nitrous acid, but the purine product, i.e., xanthine is unable to pair either with thymine or cytosine, and thus lead to inactivation of DNA or to lethality.
4. Alkylation:
Sulfur containing mustard gases, ethyl methane sulfonate etc. are called alkylating agents, as they donate an alkyl group to amino or keto groups in nucleotides.
These may produce mutations in at least three ways:
(i) Addition of methyl or ethyl groups to guanine, causing it to behave as a base analogue of adenine and thereby producing pairing errors (Fig. 3.79);

(ii) Loss of alkylated guanine bases (depurination), producing gap in the DNA chain which may interfere with DNA replication leading to chromosome breakage; and
(iii) Cross linkage between the strand of the same or different DNA molecules causing the loss or excision of nucleotides.
In addition to the above mentioned mutagens, hydroxylamine is a potent mutagen that can add a hydroxyl group to the amino group of cytosine. Thus, it enables the latter to undergo a tautomeric shift, so that it can pair with adenine (Fig. 3.80).

(b) Frame Shift Mutation:
Mutations that result from the addition or deletion of one or more base pairs in a gene are called frame shift mutations. Such a change will cause- all the subsequent three-letter words (codon) to be changed, i.e., the frame of reading in mRNAs will be altered’. A frame shift mutation will occur when any number of bases are added or deleted except in multiples of three, which would re-establish the initial frame of reading.
Cause of Frame Shift Mutation:
Acridine dyes, such as pro-flavin and acridine orange, ithedium bromide, ICR-170 etc. are mutagens that can cause frame shift mutation. They are of roughly the same dimensions as a nitrogenous base pair and are known to intercalate or wedge between the purines and pyrimidine’s of intact DNA, inducing contortions in the DNA helix and causing deletions and additions.
During replication, there is a possibility of slippage and improper base-pairing of one strand with the other. The intercalation of the acridine into an improperly base-paired region can extend the existence of these slippage structures and that will ultimately result in an addition or deletion of one or more bases in one of the strands.
Examples of Point (Gene) Mutation in Man:
Sickle Cell Anaemia:
The order of nucleotides in DNA specifies the order of amino acids in a protein. Mutations in a gene would correspond to alterations in the sequence of DNA, which might result from, the substitution of one nucleotide for another or from the addition or deletion of nucleotides.
These changes in the nucleotide sequence of DNA would then lead to corresponding changes in the amino acid sequence of the protein encoded by the gene in question. So mutations within a single gene could alter different amino acids in the encoded protein, and that altered protein may sometimes cause important physiological disorders. Such a gene mutation is found in patients with sickle-cell anaemia.
Sickle-cell anaemia is an autosomal disease that results from the substitution of a single amino acid in the haemoglobin protein that transports oxygen in the blood. In 1910, J. Herrick found that red blood cells from individuals with this disease contain erythrocytes that, under low oxygen tension, become elongated and curved because of the polymerization of haemoglobin.
The “sickle” shape of these erythrocytes is in contrast to the biconcave disc shape of normal individuals (Fig. 3.81a). This disease is almost exclusively ‘found in the African-American population where it affects about one in every 625 infants.

Symptoms of Sickle-cell Anaemia:
The sickled red blood cells are more fragile than normal blood cells and tend to break easily. In addition, sickled cells clog the capillaries rather than squeeze through them. So, individuals with sickle-cell anaemia, suffer attacks when red blood cells aggregate in the venous side of capillary system, where O2 tension is very low.
As a result, a variety of tissues may be deprived of O2 and suffer severe damage. The kidneys, heart, muscles, joints, brain, gastrointestinal tract and lungs may be affected. If untreated, the affected individuals may therefore suffer from a variety of health problems, including heart failure, pneumonia, paralysis, kidney failure, abdominal pain and rheumatism.
Compensatory physiological mechanisms include red-cell production by bone marrow and accentuated heart action. These mechanisms lead to abnormal bone size and shape as well as dilation of the heart.
Haemoglobin in Sickle-cell Anaemia:
Haemoglobin consists of four polypeptide chains — two α polypeptides and two β- polypeptides — each of which is associated with a haem group. Sickle-cell anaemia is caused by homozygosity for a mutation in the p-globin gene which encodes the β- polypeptide of haemoglobin.
The sicklercell mutation βs is co-dominant with the wild type allele βA. The heterozygous βAβS produces two types of haemoglobins. The normal HbA has two normal α-chains and two normal β-chains.
The other, called Hbs, is the defective haemoglobin, with two normal α-chains and two abnormal β-chains specified by the mutant p-globin gene βs.
These βAβs heterozygotes are called sickle-cell trait, they usually show few symptoms of the disease but sickling of red cells may occur, giving rise to symptoms similar to affected persons following sharp drop in O2 tension. Affected individuals result from the homozygous genotype Hbs//Hbs.
Molecular Basis of Sickle-cell Anaemia:
In 1950s, Linus Pauling and his co-workers first provided the insight into the molecular basis of the disease. They showed that haemoglobin isolated from normal individuals differ from that of haemoglobin isolated from diseased persons, in the rate of electrophoretic migration. They concluded that a chemical difference exists between normal and sickle-cell haemoglobin.
In starch gel electrophoresis, HbA migrates further than Hbs, suggesting that its net negative charge is greater. On the other hand, the electrophoretic pattern of haemoglobin derived from carriers (heterozygotes) reveals the presence of both HbA and Hbs, confirming their heterozygous genotype (Fig. 3.81b).
Pouling concluded that sickle-cell anaemia results from a mutation that alters the chemical structure of the haemoglobin molecule.
In 1957, J. Ingram demonstrated that chemical changes occur in the primary structure of the globin portion of the haemoglobin molecule. Using finger-printing technique, Ingram showed Hbs differs from HbA in amino acid composition.
In adult human, the α-chain of haemoglobin contains 141 amino acids and β-chain contains 146 amino acids in its quaternary structure. In the β-chain of sickle- cell individuals, however, Ingram showed a single amino acid substitution at the sixth position from the N-terminal end.
It caused replacement of the acidic amino acid glutamic acid with neutral amino acid valine (Fig. 3.81c). The codon specifying glutamic acid is GAG, whereas that for valine is GUG.
Actually the base A of GAG is replaced by U. This single base pair change in the p-globin gene (A – T in the DNA is changed into T-A, therefore, in the mRNA GAG is converted into GUG) of sickle-cell person was responsible for the addition of valine that was encoded instead of glutamic acid.
This particular substitution of amino acid caused β- polypeptide to fold up in different ways. In turn, this leads to sickling of the red cells in the diseased persons. This finding provided strong evidence that a point mutation is responsible for the disease sickle-cell anaemia.
Moreover, it is established that genes specify the amino acid sequence of polypeptide chains. The a-globin and p- globin genes are located on different autosomes.
Inheritance of Sickle-cell Anaemia:
James Neel and E. A. Beet first demonstrated that this autosomal disease is inherited as a Mendelian trait. The genotypes of normal, carrier (trait) and sickle-cell anaemic individuals are HbA//HbA, HbA//Hbs and Hbs//Hbs, respectively. Marriage between carrier male and female will produce normal, sickle-cell trait and sickle cell anaemic offspring in the ratio of 1 : 2 : 1.
B. Chromosomal Mutation:
A change in the organization of a chromosome or chromosomes i.e., changes either in the number or morphology of chromosomes by deletion or duplication of genes or segments of chromosome, and (a) rearrangements of the genetic material either within or among chromosomes are called chromosomal mutations or chromosomal aberrations (Table 3.8).
As chromosome is the unit of genetic transmission so chromosome aberrations are passed on to offspring in a predictable manner, producing many heritable phenotypic variations.

(a) Variation in Chromosome Number:
Number of sets of chromosome of any organism is called ‘ploidy’ Mutations involving chromosome number may be of two types:
(i) Aneuploidy:
Mutations which involve changes in the number of any single chromosome within a set.
It is again of several types:
1. Monosomy:
The loss of a single chromosome from an otherwise diploid genome is called monosomy. The loss of one chromosome produces a 2n-l complement. The monosomic aneuploidy will produce two types of gametes, (n-1) and (n). The (n-1) gamete when fertilized by normal (n) gamete will again produce (2n-l) or monosomic offspring. In man monosomy of one X chromosome produces Turner’s syndrome.
Example of monosomy in man:
Turner Syndrome:
In 1938, Turner observed some human females with sexual infantilism. Later Wilkins and Fleischmann (1944) described ‘streak’ gonads devoid of ovarian follicles in such females. Pollani et al. (1954) and Wilkin et al. (1954) demonstrated that most of such females are chromatin negative whereas Ford et al. (1956) described an XO karyotype in such females.
Such females are called Turner females. About 1 in 3,000 female births show Turner syndrome. They show the following characteristics.
Characteristics of Turner Females:
1. Turner females have female like external genitalia but are nevertheless chromatin- negative. Their ovaries do not develop properly and are represented by fibrous streaks. Primordial follicles are usually absent. Uterus is small.
2. Underdeveloped breast consisting mostly of fat.
3. Short stature (usually under 5 feet); broad, shield-like chest with widely spaced nipples.
4. Webbed neck and a low posterior hairline is seen.
5. Lymphedema of dorsum of the hand and feet.
6. Nails are small, narrow and set deep into the nailpit or are square with increased lateral curvature.
7. Intellectual impairment is occasionally present.
8. Colour blindness, diabetes mellitus and congenital pyloric stenosis are higher in Turner female than normal woman.
Origin:
The most common chromosomal constitutions in Turner female is 44 autosomes and only one X-chromosome. They are designated 45, X (Fig. 3.83). They have no Barr-body.
The XO constitution may arise by following ways:

i. Monosomy of X-chromosome:
Monosomy of X-chromosome occurs in human due to non-disjunction or loss of one X- chromosome during meiosis (Fig. 3.82) (non-disjunction, see sex determination Page 353). Due to the absence of a Y- chromosome, no musculinization occurs in Turner syndrome.
 ii. Mosaicism:
ii. Mosaicism:
Turner syndrome can also result from karyotypes other than 45, X. Individuals called mosaics whose somatic cells display two different genetic cell lines, each exhibiting a different karyotypes. The most common chromosome combinations being 45, X/46, XY and 45, X/46, XX. Such cell lines result from a mitosis error during early development.
So, the embryos beginning life with a normal karyotype, can give rise to an individual whose cells show a mixture of karyotypes and expressing this syndrome. Partial deletion of the short arm of X chromosome and of long arm (Gacobs et al., 1960, 1961) has been reported in a few Turner females.
However, Pollani (1961, 1962) has reported chromatin positive females with neck webbing and other anomalies of Turner syndrome that have normal ovarian function and normal XX karyotype.
2. Partial Monosomy:
Autosomal monosomy has not been reported beyond birth in human. There are, however, survivors with partial monosomy, where only part of one chromosome is lost. These cases are also called segmental deletions.
Example of partial monosomy in man:
Cri-du-chat Syndrome:
In 1963, Jerome Le Jene discovered individuals (1 in 50,000) where one-half of the short arm of chromosome 5 is lost. Their genetic constitution is designated as 46, -5p, meaning that all 46 chromosomes are present, but some or all of the p arm (the short or petite arm) of chromosome 5 is missing (Fig. 3.84).
Individuals with such chromosome constitution exhibit some syndrome referred to as ‘Cri-du-chat’ syndrome (‘cry of a cat’). The size of the deletion appears to influence the physical, psychomotor and mental skill levels of these individuals.

Characteristics:
i. Individuals with cri-du-chat syndrome show abnormal development of glottis and larynx. As a result, the infant has a cry similar to that of meowing of cat, thus giving the syndrome its name.
ii. Malformed anatomy with gastrointestinal and cardiac complication.
iii. Mental retardation are common in the affected individuals. However, many individuals achieve a level of social development in the trainable range.
iv. Unique facial features include widely spaced eye with epicanthic folds.
Origin:
A partial, deletion may result when the short arm of chromosome 5 is trans-located to a chromosome of another group (e.g., 15 in the D group). Although this person is normal, he or she will nevertheless produce some unbalanced gametes which carry only the deficient member of the translocation pair. Children inheriting this deletion gamete will show the cri-du-chat syndrome.
3. Double Monosomy:
When two different or non-homologous chromosomes are lacking from the diploid set of chromosome. The genomic formula will be (2n-1-1).
4. Nullisomy:
When a pair of homologous chromosomes are missing from the diploid set of chromosome, it is called nullisomy. The genomic formula will be (2n-2). The gametes produced will be (n-1) and (n-1). Sears could produce 21 types of nullisomy in wheat.
5. Trisomy:
When an extra chromosome is present in addition to the normal diploid condition, trisomy is formed. The genomic formula will be (2n+1). The gametes thus formed will be (n+1) and (n). For example, in man trisomy of chromosome 21 produces Mongolism or Down’s syndrome.
6. Double Trisomy:
When two non-homologous chromosomes are added to- the normal diploid member of chromosomes double trisomy results. The genomic formula will be (2n+1+1).
7. Tetrasomy:
Tetrasomy is formed when a homologous pair of chromosome is added to the normal diploid set. The genomic formula will be (2n+2). The gamete formula will be (n+1) and (n+1). Tetrasomies have been identified in wheat.
Example of trisomy in man:
Down Syndrome:
In 1966, Langdon Down described the only human autosomal trisomy that occurs in the chromosome 21 i.e., each cell contains three chromosomes of 21 rather than two. This condition is called Down syndrome or trisomy 21 or Mongolism due to their facial characteristics very similar to that of the Mongolian race. It is probably the most common congenital abnormality, with a frequency of one out of 600 births.
Physical and physiological characteristics:
i. Individuals with Down syndrome have a prominent epicanthic fold in the corner of each eye.
ii. They are short with dull and happy looking, short hands with fingers showing characteristic palm and finger print patterns and feet with wide gap between first and second toes.
iii. They may have flat, round heads; protruding furrowed tongues which cause the mouth to remain partially open.
iv. Both physical and mental development is retarded and poor muscle tone is characteristic.
v. Their life expectancy is shortened, although individuals, are known to survive into their 50s.
vi. No sexual maturity. Testes are small and undescended in male and in females labia majora tend to be large while labia minora are small or absent.
vii. Down children are prone to respiratory disease and have malformed heart. They show an incidence of leukemia, about 15 times higher than that of normal population.
viii. The death of older Down syndrome adults, is strikingly due to Alzheimer’s disease.
Origin of Down syndrome:
1. Trisomy of Chromosome 21:
Nondisjunction of chromosome 21 during either anaphase I or II of meiosis in male and female can result in the production of gametes with chromosome composition of n+1. These gametes when fertilized by a normal gamete, the trisomic condition is created.
It has been found that the ovum is the source in about 95% of these trisomes. This condition was supported by the evidence of direct relationship between the incidence of Down birth and maternal age. For example, the frequency is about 1 in 1000 at maternal age 30. A 10-fold increase is noted at age 40 having a frequency of 1 in 100.
The frequency increases further to about 1 in 50 at age 45. Why the non-dis-junctional event that produces Down syndrome seems more likely to occur during oogenesis in women between the ages of 35 to 45, is not yet known. However, age of ovum is thought to be one of the causes of the increased incidence of nondisjunction, leading to Down syndrome.
2. Translocation:
Familial Down Syndrome:
In rare cases, the occurrence of Down syndrome is expected to run in a much higher frequency over several generations. These instances are referred to as familial Down syndrome, involving a translocation of chromosome 21.
Analysis revealed that one of the parents of such down child contains a 14/21 D/G translocation. That is, one parent has the majority of the G-group chromosome 21 trans-located to one end of the D- group chromosome 14. This individual is normal even though he or she has only 45 chromosomes.
During meiosis, one fourth of the individual’s gametes will have two copies of chromosome 21 — a normal chromosome and most of a second copy trans-located to chromosome 14. When such a gamete is fertilized by a standard haploid gamete, the resulting zygote has 46 chromosomes but three copies of chromosome 21. These individuals show Down syndrome.
(ii) Euploidy:
Mutations that involve changes in the number of entire set of chromosome are called euploidy.
1. Haploidy:
It is when a cell contains only one set of chromosome. The genomic formula is (n), e.g., gametes of diploid organisms; male honey bee is haploid and develops parthenogenetically. Haploids are less vigorous than diploid prototypes.
2. Diploidy:
It is when two sets of chromosomes occur in a cell of an organism, e.g., most of the higher animals and plants. Genomic formula is (2n). They have normal growth rate, fertility and viability.
3. Polyploidy:
When more than two sets of chromosomes are present per cell of an organism is called polyploidy. When three sets occur it is called triploidy (3n); when four sets it is tetraploidy (4n); 5 sets is pentaploidy (5n); 6 sets is hexaploidy (6n); 7 sets is heptaploidy (7n); 8 sets is octaploidy (8n) and so on. Polyploidy is rare in animal kingdom.
A polyploidy disturbs the sex determination mechanism. However, polyploidy has been reported in Daphnia, Ascaris, silk-moth etc. In plants, polyploidy is very common, e.g., cotton, wheat, tobacco, rose etc. have different types of polyploid varieties.
Polyploidy can be classified once again into auto-polyploidy and allopolyploidy. When the sets of chromosome in a polyploid are derived from the same species, it is known as auto-polyploidy.
For example, if an exceptional diploid gamete is fertilized by a normal haploid gamete, triploid will be produced. Again if the chromosome number in a diploid is doubled due to failure of spindle formation, tetraploid will be produced e.g., seedless autopolyploid grapes, banana etc.
When a polyploid is produced from two different species, it is called as allopolyploidy. For example, two species are hybridized and then the chromosome number of hybrid is double, thus allotetrapolyploid is produced. For example, Raphanobrassica is an allotetraploid between Rhaphanus (radish) and Brassica (mustard), but unfortunately this plant has root-like mustard plant and shoot-like radish plant.

(b) Variation in Structure of Chromosome (Structural Aberration):
Variation in the structure of chromosomes include:
(i) Deletion or Deficiency:
Loss of a part of chromosome,
(ii) Duplication:
Addition of a part of chromosome
(iii) Inversion:
Rearrangements of genes in which a chromosome segment is inverted and
(iv) Translocation:
Exchange of segments between non-homologous chromosomes (Fig. 3.86).

In most instances, these structural changes are due to one or more breaks along the axis of a chromosome, followed by either the loss or rearrangement of genetic material. The breakage is spontaneous, but the rate of breakage may increase in cells exposed to chemicals or radiation.
If breakage and rejoining does not reestablish the original relationship, and this alteration occurs in germplasm, then the gametes will contain a heritable structural rearrangement. Structural abnormalities may be found in both the homologous chromosomes of a pair, or in only one of them. The first case is called structural homozygotes and the latter type is called structural heterozygotes.
(i) Deletions/Deficiencies:
If a chromosome breaks in one or more places and a portion is lost, the missing piece is referred to as a deletion or a deficiency. Deletion occurring near one end of the chromosome is called terminal deletion and that which results from the interior of the chromosome is called intercalary deletion (Fig. 3.87a, b).
Part of the chromosome that retains the centromere will divide normally, whereas the segment without centromere will eventually be lost in progeny cells following mitosis or meiosis.
Synapsis that occurs between a chromosome with large intercalary deficiency and a normal complete homologue, the unpaired region of the normal homologue must ‘buckle out’ into a loop called ‘deficiency or compensation loop’ (Fig. 3.87c).
In human, only a part of the short arm of chromosome 5 is lost and results in the cri-du-chat syndrome. Deletions have an effect on inheritance also. In presence of a deficiency, a recessive allele will behave like a dominant allele, a phenomenon known as pseudo-dominance, e.g., notch phenotype in Drosophila.

(ii) Duplication:
When a single locus or a large piece of a chromosome is present more than once in the genome, it is called duplication. Like deletion, duplication may also produce a compensation loop following synapsis.
Duplication can arise as the result of unequal crossing over between synapsed chromosomes during meiosis or through a replication error prior to meiosis (Fig. 3.88). Duplication may result in three important phenomena.

1. Gene Redundancy:
It is a phenomenon which represents the presence of multiple copies of a gene, coding for a single RNA, e.g., rDNA coding for rRNA.
2. Phenotypic Variation:
Duplication may cause phenotypic variations that at first might appear to be caused by a simple gene mutation. The best example is bar eye phenotype in Drosophila. Bar eyed flies have narrow, slit-like eyes. This phenotype appears to be inherited as a dominant X-linked mutation.
However, studies of C. Bridges and J. Muller revealed that one copy of region 16A of the X-chromosome is present in wild- type flies. This region is duplicated in bar flies and triplicated in double bar flies. So bar phenotype results due to duplication but not from a simple chemical change in the gene.
The double bar condition originates as a result of unequal crossing- over. So all these altered phenotypes result from new ‘positioning’ of a gene within the genome. This is referred to as position effect or dosage effect.
3. Genetic Variation:
When an essential gene becomes duplicated in a germ cell, major mutational changes result in the organism. These extra changes would be tolerated by the organism because the original gene still provides the genetic information for its essential function.
However, over a long evolutionary period, the duplicated gene may change sufficiently so that its product assumes a divergent role in the cell. Thus, the new function may impart an adaptive advantage to the organisms, enhancing their fitness.
(iii) Inversion:
Inversion is a type of chromosomal aberration in which two breaks occur in a chromosome and the intercalary segment reunites in reverse order i.e., the segment rotates at 180°. Inversion never involves loss of genetic information, but simply rearrangement of linear gene sequences. The inverted segment may be short or quite long and may or may not include the centromere.
If the centromere is included in the inverted segment, it is called pericentric inversion (Fig. 3.89). If the centromere is not included in the inverted segment, it is called paracentric inversion (Fig. 3.89). The ratio of arm lengths extending from the centromere remain unchanged in paracentric inversion (Fig. 3.89). In contrast, pericentric inversions may create chromosomes with arms of different lengths.
Inversions seem to have minimal impact on the genetic material but their consequences are interesting to geneticists because inversions may result in position effects and might play an important role in evolutionary process.

Normal linear synapsis during meiosis is not possible when only one member of a homologous pair of chromosome has an inverted segment. In order to enable pairing between two such chromosomes, a shape of loop (inverted loop) is formed by each of the two chromosomes as shown in Fig. 3.90.
In other cases, if no loop is formed, the homologues are seen to synapse everywhere except along the length of the inversion, where they appear separated.

If crossing over does not occur within the inverted segment of the inversion heterozygote, the homologues will segregate and result in two normal and two inverted chromatids that are distributed into gametes, and the inversion will be passed on to one-half of the offspring (Fig. 3.90).
However, if crossing over occurs within the inversion loop, abnormal chromatids are produced. A single or odd number of cross-overs in inverted region of paracentric inversion result into the formation of a dicentric chromosome (with two centromeres) and an acentric (with no centromere) chromosome (Fig. 3.91).
These chromatids will be observed at anaphase I in the form of a bridge and a fragment. This structure is referred to as dicentric bridge (Fig. 3.92). Double crossovers or crossovers of even number within and outside the inversion will give various kinds of deficiencies and duplications.


In case of pericentric inversion, similar chromosomal imbalance is produced as a result of a crossover event between a chromatid bearing a pericentric inversion and its non-inverted homologue (Fig. 3.93). However, no acentric and dicentric chromatids are produced. The recombinant chromatids that are directly involved in the exchange have duplications and deletions.
The gametes having these chromosomes will not function. Since the products of single crossover will not function and the only crossovers recovered will be double cross-overs, the observed frequency of recombination between any two genes in question will be considerably reduced.
Due to this reason, inversions are often called crossover suppressors. This reduction in crossing over is not the actual reduction in cytological crossing over, but is the result of lack of recovery of the products of single crossovers. Because of this suppression of the recovery of crossover products, a particular combination of alleles is preserved intact in the viable gametes.
If the allele of the involved genes provide a survival advantage to organisms maintaining them, the inversion will be beneficial to the evolutionary survival of the species. For example, maintenance of different inversions on chromosome 3 of Drosophila pseudoobscura through many generations has been proved to be highly adaptive to this species.

(iv) Translocation:
Translocation involves movement of a chromosomal segment to a new location in the genome. Reciprocal translocation involves the exchange of segments between two non-homologous chromosomes. Like inversions, in translocation there is no gain or loss of genetic information, rather, there is only a rearrangement of genetic material.
If a translocation is present in one of the two sets of chromosomes, it will be a translocation heterozygote. In these cells, normal pairing into bivalents will not be possible among chromosomes involved in translocation.
However, pair between homologous segments of chromosomes results in a cross- shaped (+) figure, involving four chromosomes, observable at the pachytene stage. In case of translocation, however, aberrant gametes are not necessarily the result of crossing over.
According to the principle of independent assortment the chromosome containing centromere 1 will migrate randomly towards one pole of the spindle, during the first meiotic anaphase; and will travel with the chromosome either having centromere 3 or centromere 4.
The chromosome with centromere 2 will move to the other pole along with the chromosome containing either centromere 3 or centromere 4. This results in four potential meiotic products of which 1, 4 combination contains chromosomes uninvolved in the translocation and the 2, 3 combination contains trans-located chromosome.
The other two potential products, the 1, 3 and 2, 4 combinations, will contain chromosomes displaying duplicated and deleted segments (Fig. 3.94 a, b and Fig. 3.95).
If the resultant meiotic products participate in fertilization, lethality often results. As few as 50 percent of the progeny of parents heterozygous for a reciprocal translocation may survive. This condition is called semi-sterility that has an impact on the reproductive fitness of organisms, thus playing a role in evolution.


In human, such abnormal condition leads to variety of birth defects like familial Down syndrome, fragile X-syndrome etc. Ninety percent of chronic myelogenous leukemia (CML) patients have a chromosomal mutation in the leukemic cell called the Philadelphia chromosome (Ph’).
It results from a reciprocal translocation event in which a part of the long arm of chromosome 22 is trans-located to chromosome 9, and a small part from the tip of chromosome 9 is trans-located to chromosome 22. Similarly 90% of tumors Burkett’s Lymphoma are associated with a reciprocal translocation involving chromosomes 8 and 14.
Type # 2. According to Direction:
A. Forward and B. Reverse mutation:
Forward mutations are mutations that cause the genotype to change from wild to mutant type. Mutations that cause the genotype to change from mutant to wild type is called reverse or backward or back mutation.
This is of four types:
A. Single Site Mutation:
One nucleotide changes into another nucleotide and the mutated nucleotide changes back into the original.
B. Photo-reactivation:
In presence of light waves, photo-reactivation enzyme can cleave the thymine-dimer produced by UV radiation.
C. Dark Reactivation:
Endonuclease may remove the dimer which is digested by exonuclease and missing part is correctly synthesized by DNA polymerase and attached to the polynucleotide chain by ligase.
D. Mutation Suppressor:
A mutation at another site sometimes corrects the effects of the primary mutation.
Type # 3
. According to Magnitude and Phenotypic Effect:
According to the magnitude of phenotypic effect, mutation is of three types:
A. Dominant Mutation:
The mutation that can express over the wild type is called as dominant mutation, so that they can express in heterozygous condition, e.g., defective tooth enamel in man is due to a X-linked dominant mutation.
B. Recessive Mutation:
The mutation that cannot express over the wild type is called recessive mutation, which can express only in homozygous state e.g., black body in Drosophila.
C. Lethal Mutation:
Lethal mutation is the mutation that causes the death of any organism either during development or in adult stage, but before the attainment of sexual maturity. Complete lethal mutation causes cent percent mortality, semi-lethal causes more than 90% but less than 100% and sub-vital causes more than 10% but less than 90% mortality.
Type # 4
. According to Origin:
According to the mode of origin, mutation is of two types:
A. Spontaneous and B. Induced mutation. Random changes in nucleotide sequences of a gene are called Spontaneous mutation. No specific external agents are associated with their occurrence. Most spontaneous mutations occur during the enzymatic process of DNA replication which leads to error in the genetic code.
The error is then reflected in the amino acid composition of the protein which in turn is reflected in the protein’s function. Mutations that result from the influence of any external or artificial factors are considered to be induced mutations. Any physical and chemical agent that significantly increases the frequency of mutational events above a spontaneous maturation rate is called mutagen.
Radiations, both ionizing (e.g., X-ray) and non-ionizing (e.g., UV light) are used to induce mutations. Collision of ionizing radiation with atoms in its path gives rise to ions and reactive chemical radicals that break chemical bonds, including those in DNA.
Thus, the products of ionizing radiation can induce chromosome breakage, chromosome rearrangements and damage to DNA, e.g., point mutation. In human, the effects of ionizing radiation doses are cumulative.
That is, if a particular dose of radiation results in a certain number of point mutations, the same number of point mutations will be induced whether the radiation dose is received over a short period of time or ever a long period of time.
Ultraviolet light or UV ray are nonionizing radiation, that is, they have insufficient energy to induce ionization. However, UV can form abnormal chemical bonds between adjacent pyrimidine molecules in the same strand or between pyrimidine’s on the opposite strands of the double helix.
This bonding is induced mostly between adjacent thymine’s, forming thymine dimers, usually designated as TᶺT (Fig. 3.96), CᶺC. CᶺT and TᶺC pairs arc also produced by UV radiation but in much lower rates. This unusual pairing produces a bulge in the DNA strand and disrupts the normal pairing of Ts with corresponding as on the opposite strand.

In normal cells, many of such thymine dimers are repaired, but if sufficient pyrimidine dimers remain unrepaired in the cell, lethal mutation may result. Rate of spontaneous mutation is exceedingly low for all the organisms studied.
The rate varies considerably among different organisms and even within the same species, the rate may vary from gene to gene. In human, the genes studied average between 1/1,000,000 and 1/100,000 mutations per gamete.
Type # 5
. According to Cell Type:
A. Gametic and B. Somatic Mutations:
Mutations occurring in somatic cells are called somatic mutation. These are not transmitted to future generations. These have no importance in evolution. Somatic mutations will have a greater impact only if they are dominant and occur early in development.
Dominant autosomal mutations will be expressed phenotypically in the first generation while autosomal recessive mutations in the gametes of either males or ‘females may go unnoticed for many generations until the mutant allele has become widespread in population.
Radiation in many somatic tissues causes chromosomal aberration. Gametic mutation or mutation occurring in gametes has greater significance for evolution, because as a part of germ line, they are transmitted to offspring’s, e.g., red green colour blindness in man.
Type # 6
. Other Types of Mutation:
A. Missense and Nonsense Mutations:
In a missense mutation, a base pair change in the DNA causes a change in an mRNA codon so that a different amino acid is inserted into the polypeptide chain in place of the one specified by the wild- type codori, resulting in an altered phenotype.
For example, in human, single nucleotide pair change in codon 6 of the β-haemoglobin gene leads to an amino acid substitution in the β-haemoglobin chain. If the individual is homozygous for this mutation, he or she will develop sickle-cell anaemia.
A nonsense mutation involve a change in base pair in the DNA that results in the change of an mRNA codon to one that specifies an amino acid to a chain-terminating (nonsense) codon, viz., UAG, UAA or UGA.

B. Morphological mutation:
Mutations that affect some morphological traits and are recognized on the basis of their deviation from the normal or wild type- phenotypes are called morphological mutation. All of Mendel’s pea characters were morphological mutations.
C. Nutritional or Biochemical Mutation:
These are mutations involving biochemical or nutritional variations. These may not affect specific morphological characters. In human, haemophilia is an example of biochemical mutation.
D. Behaviour Mutation:
Mutations may also affect an organism’s behaviour pattern and are therefore called behaviour mutation. For example, mating behaviour or circadian rhythm of animal can be altered.
E. Regulatory Mutation:
Here the mutation of one gene may control the function of another gene, e.g., lac operon.
Detection of Mutation:
Geneticists apply different genetic techniques, cell cultures and pedigree analysis to detect different mutations in different organisms. H. J. Muller developed a number of systems for detecting and estimating the spontaneous and induced rates of X-linked and autosomal recessive lethal mutations in Drosophila.
In Muller’s classical CIB technique, wild type male flies are treated with a mutagenic agent X-rays and are mated to untreated, heterozygous CIB females (one X-chromosome has three marker genes, viz., C, an inversion that suppresses the recovery of crossing-over products, called ‘cross over suppressor’; l, a recessive lethal allele; and B, the dominant gene duplication causing bar eye).
In the F, generation, females expressing bar eye are selected. They have received one X from their mother (the CIB chromosome) and one X from their father (Fig. 3.97). Some of the parental X-chromosomes will bear an induced X-linked lethal mutation.

When an F, bar female that is heterozygous for such a lethal mutation is then backcrossed to a wild- type male, no male progeny will result. Half of them die because they are hemizygous for the CIB chromosome (l is lethal), and the other half die because they are hemizygous for the newly induced lethal allele.
Thus this method permits the detection of X-linked morphological mutations. Other detection method of mutation in Drosophila is the attached—X-chromosome method and pedigree analysis of haemophilia in human for X-linked mutation.