The following points highlight the five main forces that stabilise protein structures. The forces are: 1. Salt Linkages 2. Hydrogen Bonding 3. Disulfide Linkages 4. Hydrophobic Interactions 5. Van der Waals’ Forces.
Force # 1. Salt Linkages:
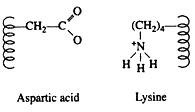
Salt linkages (ionic bonds) result from interactions between positively and negatively charged groups on the side chains of the basic and acidic amino acids. For the mutual attraction between an aspartic acid carboxylate ion and a lysine ammonium ion helps to maintain a particular folded area of the protein:
Force # 2. Hydrogen Bonding:
Hydrogen bonds are formed principally between the side chains of the polar amino acids and between a carboxyl oxygen and a hydrogen donor group. Hydrogen bonds (as well as salt linkages) are extremely important in the interaction of protein with other molecules:
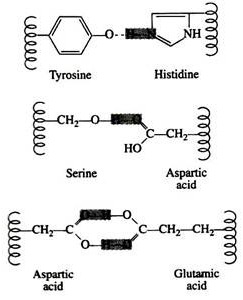
A significant feature of hydrogen bonds is that they are highly directional. The strongest hydrogen bond results when the hydrogen donor and the acceptor atom are co-linear. If the acceptor atom is at an angle to the covalently bonded hydrogen atom, the hydrogen bond is much weaker:
Force # 3. Disulfide Linkages:
Two cysteine residues may come in proximity as the protein molecule folds. The disulfide linkage results from the subsequent oxidation of the highly reactive sulfhydryl (—SH) groups to form cysteine:
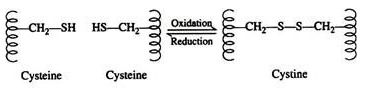
This disulfide bridge is the second-most important covalent interaction involved in protein structure. Disulfide linkages are frequently found in proteins as a general aid to the stabilization of the tertiary structure. Note, that one or more of these bonds may join one portion of a polypeptide chain covalently to another, thus interfering with the helical structure.
Force # 4. Hydrophobic Interactions:
ADVERTISEMENTS:
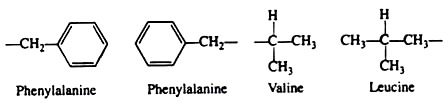
Many investigators now believe that the non-covalent hydrophobic forces are the most significant in stabilising the conformation of a polypeptide chain. It is not because they are so strong, but rather because there are so many of them. The majority of the nonpolar amino acid groups, cluster together at the interior of the chain and the strength of all their hydrophobic interactions is considerable:
Force # 5. Van der Waals’ Forces:
These are extremely weak forces and act only over extremely short distances; include both an attractive and a repulsive component. The attractive force involves interaction between induced dipoles formed by momentary fluctuations in the electron distribution in nearby atoms.
ADVERTISEMENTS:
The repulsive force comes into play when two atoms come so close that their electron orbitals overlap. The distance at which the attractive force is maximal and the repulsive force is minimal is termed the Van der Waals’ contact distance. Atoms have characteristic Van der Waals’ radii and the optimal contact distance between two atoms is the sum of their Van der Waals’ radii.
α-helix and p-pleated sheet of Secondary structures:
The regular folding of regions of the polypeptide chain is the feature of the secondary level structure of protein. These foldings are commonly of two types – α-helix and β-pleated sheet. In the rod-like α-helix, the amino acids arrange themselves in a regular helical conformation (Fig. 8.70a). The carbonyl oxygen of each peptide bond is hydrogen bonded to the hydrogen on the amino group of the fourth amino acid away (Fig. 8.70b).

These hydrogen bonds running nearly parallel to the axis of the helix. In an α-helix there are 3.6 amino acids per turn of the helix covering a distance of 0.54 nm. The side chains of the amino acids are all positioned along the outside of the cylindrical helix (Fig. 8.70c). The distance between two consecutive α-carbons of amino acids is 0.15 nm.
Hydrogen bonds are forming between different polypeptide chains or between two distant peptide bonds of the same polypeptide in case of β-pleated sheet (Fig. 8.70d). Adjacent polypeptide chains in β-pleated sheets can be either parallel or antiparallel (Fig. 8.70e).
The polypeptide chain within a β-pleated sheet is fully extended, and the distance between two Cα of adjacent amino acids in 0.35 nm. β-pleated sheets are always slightly curved and, if several polypeptides are involved, the sheet can close up to form a β-barrel.
The polypeptide chain often reverses its direction, making a hairpin or β-turn, for compact and tight folding in a globular protein. In β-turn the carbonyl oxygen of one amino acid is hydrogen bonded to the hydrogen on the amino group of the fourth amino acid (Fig. 8.70d). Regions of the polypeptide chain that are not in a regular secondary structure are said to have a coil or loop conformation.

The linkages responsible for tertiary structure are formed between side chains of the amino acids. Table 8.11 gives an indication of the relative strengths of interactions involving the non-covalent bonds found in proteins.
